

版納園以自主開發(fā)的比較基因組程序包對174個葉綠體全基因組進行分析
高通量測序(High-throughput sequencing),又稱“下一代”測序("Next-generation" sequencing),是近年來在測序技術發(fā)展史中具有革命性改變的新突破,能一次并行對幾十萬到幾百萬條DNA分子同時測序,因此能對物種的轉錄組和基因組進行比以往較細致全貌的分析。但是,由于“下一代”測序技術原始數(shù)據(jù)的讀長(read length)只有幾十個或一、兩百個堿基,按照傳統(tǒng)的分析流程,必須要通過生物信息學工具將這些短的堿基數(shù)據(jù)組裝成較長的序列組(contigs)或基因組的框架,或者把這些序列比對到已有的參照基因組或者相近物種基因組序列上,才能進一步取得具有生物學意義的結果。對于沒有參照基因組的非模式生物,要把這些海量的短序列數(shù)據(jù)組裝的工作面臨一定程度上的難度,制約了這類數(shù)據(jù)在非模式生物基因組研究的發(fā)展。
考慮到大部分生態(tài)學研究里的熱帶生物都是沒有參照基因組的非模式生物,在版納植物園生態(tài)進化組Cannon研究員的領導下,版納植物園、北京基因組所及德州理工大學的科研人員研發(fā)了直接分析高通量短序列數(shù)據(jù)的程序包,簡化了高通量數(shù)據(jù)的比較基因組和轉錄組研究。由于此方法不需事先組裝基因組,而以直接通過分析檢測數(shù)據(jù)中的kmer片段是否存在及其出現(xiàn)頻次,來探討一定數(shù)量目標基因組中的序列差異,所以可以突破此類數(shù)據(jù)經(jīng)常面臨的生物信息學的分析瓶頸。通過篩選單個基因組獨有或多個基因組共享的kmer片段及找出含這群kmer片段的數(shù)據(jù)后,此程序可以對這些數(shù)據(jù)進行組裝,以取得較長的序列探討下一步的生物學問題。
基于先前的工作基礎(見已在Molecular Ecology 發(fā)表的論文,CANNON, C. H., KUA, C.-S., ZHANG, D. and HARTING, J.R. (2010), Assembly free comparative genomics of short-read sequence data discovers the needles in the haystack. Molecular Ecology, 19:147–161),我們進一步改善了非組裝分析法,以比較174個葉綠體全基因組數(shù)據(jù)印證此程序包的功能和運行流程,并于PlOS ONE發(fā)表了題為Reference-Free Comparative Genomics of 174 Chloroplasts的論文。由于這174個由低等植物和高等植物組成的葉綠體全基因組分析涉及的內容十分廣泛,我們只能簡潔的闡述幾個發(fā)現(xiàn),如雖然植物葉綠體基因組的基因結構和含量看起來十分保守,但是kmer片段分析可以把不同支流的植物清楚的分類。寄生植物的葉綠體基因組表現(xiàn)出預期的整體進化加速,而半寄生植物比全寄生植物的葉綠體基因組中含有較多的新基因序列,印證了基因組的演化機制受控于其功能。我們也發(fā)現(xiàn)了一段在被子植物里非常保守的基因序列。這分析里所有的成果都在該文章的補充材料部分。
此程序包內有4個不同功能的程序,可用LINUX和蘋果操作系統(tǒng)以命令行運行。程序包已上傳到全球最大開源軟件開發(fā)平臺sourceforge,下載網(wǎng)址為:http://sourceforge.net/projects/referencefree/
此研究得到了中國科學院知識創(chuàng)新工程重要方向項目和云南省高端科技人才引進計劃項目的資助。
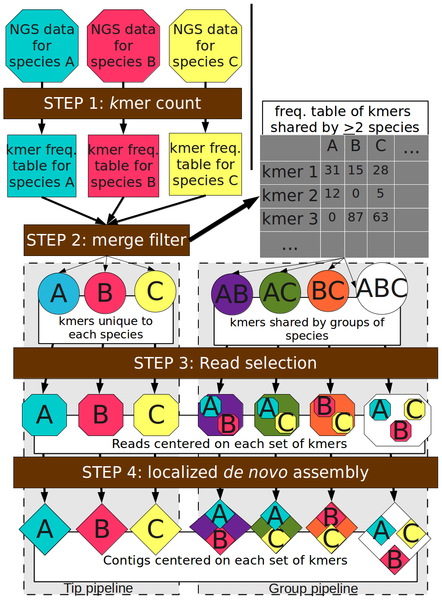
非組裝比較基因組分析流程



滇ICP備13004273號 滇公網(wǎng)安備53282302000011號


今日頭條

官方微博

微信公眾號
官方抖音號